

Structure du glycogène, synthèse, dégradation, fonctions
- 2717
- 442
- Eva Henry
Il Glycogène C'est le glucides de stockage de la plupart des mammifères. Les glucides sont communément appelés sucres et ceux-ci sont classés en fonction du nombre de déchets causés par l'hydrolyse (monosaccharides, disaccharides, oligosaccharides et polysaccharides))).
Les monosaccharides sont les glucides les plus simples classés en fonction du nombre de carbones contenus dans leur structure. Il y a alors les triosas (3c), les tétrosas (4c), les pentosas (5c), l'hexosous (6c), l'heptosase (7c) et les octosas (8c).
Structure chimique du glycogène montrant des liaisons glycosidiques (Source: Glykogène.SVG: Neurotoger Dérivé Travail: Marek M [Domaine public] via Wikimedia Commons) Selon la présence du groupe aldéhyde ou du groupe Cetona, ces monosaccharides sont également classés respectivement comme aldies ou ketosas.
Les disaccharides donnent naissance à l'hydrolyse, deux monosaccharides simples, tandis que les oligosaccharides produisent 2 à 10 unités de monosaccharides et de polysaccharides produisent plus de 10 monosaccharides.
Le glycogène est, du point de vue biochimique, un polysaccharide composé de chaînes ramifiées d'une aldose à six carbone, c'est-à-dire un hexose connu sous le nom de glucose. Graphiquement, il peut être représenté dans le glycogène comme un glucose. Ceci est aussi appelé l'amidon animal.
Le glucose dans les plantes est stocké sous forme d'amidon et d'animaux comme glycogène, qui est stocké principalement dans le foie et les tissus musculaires.
Dans le foie, le glycogène peut établir 10% de sa masse et 1% de la masse musculaire. Comme chez un homme de 70 kg, le foie pèse environ 1800 g et les muscles d'environ 35 kg, la quantité totale de glycogène musculaire est beaucoup plus grande que l'hépatique.
[TOC]
Structure
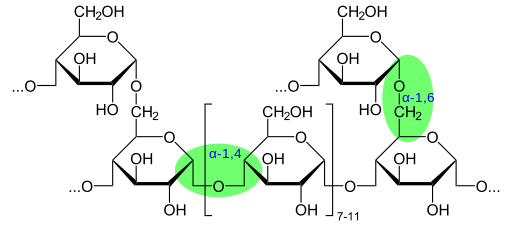
Le poids moléculaire du glycogène peut atteindre 108 g / mol, équivalent à 6 × 105 molécules de glucose. Le glycogène est composé de plusieurs chaînes α-D-glycose ramifiées. Le glucose (C6H12O6) est un aldohexosa qui peut être représenté de manière linéaire ou cyclique.
Le glycogène a une structure très ramifiée et compacte avec des chaînes de 12 à 14 déchets de glucose sous forme de α-D-glucose qui sont liés à des liaisons glucosidiques α- (1 → 4). Les ramifications de la chaîne sont formées par des liaisons glucosidiques α- (1 → 6).
Le glycogène, comme l'amidon qui est ingéré dans l'alimentation, fournit la plupart des glucides dont le corps a besoin. Dans l'intestin, ces polysaccharides sont dégradés par l'hydrolyse puis absorbés vers le torrent circulatoire principalement sous forme de glucose.
Trois enzymes: ß-amylase, α-amylase et amylo-α- (1 → 6)-glucosidase sont responsables de la dégradation intestinale du glycogène et de l'amidon.
L'α-amylase hydrolyse au hasard les liaisons α-α (1 → 4) des chaînes latérales du glycogène et de l'amidon, et reçoit donc le nom de l'endoglysidase. La ß-amyla est une exoglicosidase qui libére les dímeros ß-maltose brisant α- (1 → 4) liens glycosidiques des extrémités des chaînes les plus externes sans atteindre les ramifications.
Compte tenu du fait que ni la ß-amylase ni la α-amylase ne dégradent les branches, le produit final de son action est une structure hautement ramifiée d'environ 35 à 40 résidus de glucose qui sont appelés la dextrine limite.
La dextrine limite est enfin hydrolysée aux points de branche qui ont des liaisons α- (1 → 6) à travers l'amyle-α- (1 → 6)-glucosidase, également connue sous le nom d'enzyme "diffamatoire". Les chaînes libérées par ce débellé sont dégradées par la ß-amylase et l'α-amylase.
Alors que le glycogène ingéré entre comme du glucose, celui trouvé dans les tissus doit être synthétisé par l'organisme à partir du glucose.
Peut vous servir: purines: caractéristiques, structure, fonctionsLa synthèse
La synthèse du glycogène est appelée glycogenèse et se déroule surtout dans le muscle et le foie. Le glucose qui pénètre dans l'organisme avec le régime passe au torrent circulatoire et à l'intérieur des cellules, où il est immédiatement phosphorylé par une enzyme appelée glycoquinase.
Glucocounase phosphoryyle au glucose dans le carbone 6. L'ATP fournit du phosphore et de l'énergie pour cette réaction. En conséquence, le glucose 6-phosphate est formé et un ADP est libéré. Ensuite, le 6-phosphate de glucose devient du glucose 1-phosphate par l'action d'une phosphoglucomutase qui bouche le phosphore de la position 6 à la position 1.
Le glucose à 1-phosphate est activé pour la synthèse du glycogène, ce qui implique la participation d'un ensemble de trois autres enzymes: la pyrophosphorylase UDP-glycose, le glycogène synthétique et l'amilo- (1,4 → 1.6) -glicosyltransférase.
Le glucose-1-phosphate, ainsi que la trifosphate uridine (UTP, un nucléoside d'uridine triphosphate) et par action de l'UDP-glycose-pyrophosphorylase, forment le complexe d'uridine diphosphate-glucose (UDP GLC) (UDP GLC). Dans le processus, un ion pyrophosphate est hydrolysé.
Ensuite, l'enzyme de glycogène synthéné forme une liaison glucosidique entre le C1 du complexe GLC UDP et le C4 d'un résidu terminal de glucose de glycogène, et l'UDP UDP est libéré à partir du complexe de glucose activé par UDP. Pour que cette réaction se produise, il doit y avoir une molécule de glycogène pré-existante appelée "glycogène primaire".
Le glycogène primordial est synthétisé sur une protéine d'amorçage, la glycogénine, qui a 37 kDa et Glysila dans un résidu tyrosine en utilisant le complexe UDP GLC. De là, ils sont des déchets α-D-glucose liés avec des liaisons 1 → 4 et une petite chaîne se forme sur laquelle agit le glycogène synthésase.
Une fois que la chaîne initiale relie au moins 11 résidus de glucose, l'enzyme ramifiée ou amile- (1,4 → 1,6) -glicosyltransférase transfère une chaîne de 6 ou 7 déchets de glucose à la chaîne adjacente en position 1 → 6, qui établit une branche indiquer. La molécule de glycogène ainsi construite se développe par des ajouts d'unités de glucose avec des liens glycosidiques 1 → 4 et plus de ramifications.
Dégradation
La dégradation du glycogène est appelée glucogénolyse et n'est pas équivalente à la trajectoire inverse de sa synthèse. La vitesse de cette voie est limitée par la vitesse de la réaction catalysée par le glycogène phosphorylase.
Le glycogène phosphoryllase est responsable du fractionnement (phosphorolyse) des liens 1 → 4 des chaînes de glycogène, libérant du glucose 1-phosphate. L'action enzymatique commence aux extrémités des chaînes les plus externes et sont éliminées séquentiellement jusqu'à ce que 4 résidus de glucose restent de chaque côté des ramifications.
Ensuite, une autre enzyme, le glucano transfertas α- (1 → 4) → α- (1 → 4), laisse le point de branche exposé en transférant une unité de trisaccharide d'une branche à une autre. Cela permet à l'hydrolys Amilo- (1 → 6)-glucosidase (enzyme déstaurante). L'action combinée de ces enzymes se termine complètement en glycogène.
Comme la réaction initiale de la phosphomutase est réversible, le 6-phosphate de glucose peut être formé à partir de résidus de glucose 1-phosphate divisé à partir de glycogène. Dans le foie et les reins, mais pas dans le muscle, il y a une enzyme, du glucose-6-phosphatase, capable de rassembler le glucose 6-phosphate et de le transformer en glucose libre.
Peut vous servir: FotolyseLe glucose défier peut se propager au sang, et c'est ainsi que la glycogénolyse hépatique se reflète dans une augmentation des valeurs de glycémie (glycémie).
Régulation de la synthèse et de la dégradation
De synthèse
Ce processus est exercé sur deux enzymes fondamentales: le glycogène synthésase et le glycogène phosphorylase, de sorte que lorsque l'un d'eux est activé, l'autre est dans son état inactif. Ce règlement empêche les réactions de synthèse et de dégradation de se produire simultanément.
La forme active et la forme inactive des deux enzymes sont très différentes, et l'interconversion des formes actives et inactives de phosphorylase et de glycogène synthétique est soumise à un contrôle hormonal strict.
L'adrénaline est une hormone libérée de la moelle surrénalienne, et le glucagon en est un autre qui se produit dans la partie endocrinienne du pancréas. Le pancréas endocrinien produit de l'insuline et du glucagon. Les îlots de Langerhans α sont ceux qui synthétisent le glucagon.
L'adrénaline et le glucagon sont deux hormones qui sont libérées lorsque l'énergie est nécessaire en réponse à la diminution de la glycémie. Ces hormones stimulent l'activation du glycogène phosphorylase et inhibent le glycogène synthésase, stimulant ainsi la glycogénolyse et inhibant la glycogenèse.
Alors que l'adrénaline exerce son action sur les muscles et le foie, le glucagon n'agit que sur le foie. Ces hormones sont jointes à des récepteurs membranaux spécifiques dans la cellule blanche, qui active l'adénilate de cyclasa.
L'activation de l'adénylate cyclase commence une cascade enzymatique qui, d'une part, active une protéinquinase dépendante d'AMPC qui est inactive au glycogène synthétique et active la glycogène phosphorylase par phosphorylation (directement et indirectement, respectivement)).
Le muscle squelettique a un autre mécanisme d'activation du glycogène phosphorylase par le calcium, qui est libéré en conséquence de la dépolarisation de la membrane musculaire au début de la contraction.
De dégradation
Les cascades enzymatiques décrites ci-dessus finissent par augmenter les niveaux de glucose et lorsqu'ils atteignent un certain niveau, la glycogenèse est activée et la glucogénolyse est inhibée, inhibant également la libération supplémentaire d'adrénaline et de glucagon.
La glycogenèse est activée par l'activation de la phosphatase phosphorylase, une enzyme qui régule la synthèse du glycogène par plusieurs mécanismes, ce qui implique l'inactivation de l'inhibiteur de la kinase phosphorylase et de la phosphorylase, qui est un inhibiteur de glycogène synthésase.
L'insuline favorise l'entrée du glucose dans les cellules musculaires, augmentant les niveaux de glucose 6-phosphate, ce qui stimule la défaillance et l'activation du glycogène synthésase. Ainsi la synthèse commence et la dégradation du glycogène est inhibée.
Les fonctions
Le glycogène musculaire constitue une réserve d'énergie pour le muscle qui, comme les graisses de réserve, permet aux muscles de remplir ses fonctions. Étant une source de glucose, le glycogène musculaire est utilisé pendant l'exercice. Ces réservations augmentent avec l'entraînement physique.
Dans le foie, le glycogène constitue également une source de réserve importante pour les fonctions de l'organe et pour la contribution du glucose au reste du corps.
Cette fonction du glycogène hépatique est due au fait que le foie contient du glucose 6-phosphatase, une enzyme capable d'éliminer le groupe de phosphate de glucose à 6-phosphate et de le transformer en glucose libre. Le glucose libre, contrairement au glucose phosphorylé, peut être réparti à travers la membrane hépatocytaire (cellules hépatiques).
Il peut vous servir: sporulation: dans les plantes, dans les champignons et dans les bactériesC'est ainsi que le foie peut fournir du glucose à la circulation et maintenir des niveaux de glucose stables, même dans des conditions de jeûne prolongées.
Cette fonction est d'une grande importance, car le cerveau est nourri presque exclusivement de la glycémie, donc une hypoglycémie grave (très faibles concentrations de glycémie) peut provoquer une perte de connaissances.
Maladies connexes
Les maladies liées au glycogène reçoivent le nom générique des "maladies de stockage du glycogène".
Ces maladies constituent un groupe de pathologies héréditaires caractérisées par le gisement dans les tissus de quantités anormales ou de types de glycogène.
La plupart des maladies de stockage du glycogène sont causées par un déficit génétique de la nature de l'une des enzymes impliquées dans le métabolisme du glycogène.
Ils sont classés en huit types, dont la plupart ont leurs propres noms et chacun d'eux est produit par un déficit enzymatique différent. Certains sont mortels aux premiers stades de la vie, tandis que d'autres sont accompagnés d'une faiblesse musculaire et d'un déficit pendant l'exercice.
Exemples exceptionnels
Certaines des maladies liées au glycogène les plus importantes sont les suivantes:
- La maladie de Von Gierke ou la maladie de stockage du glycogène de type I, est produite par un déficit de glucose à 6-phosphatase dans le foie et les reins.
Il se caractérise par une croissance anormale du foie (hépatomégalie) en raison de l'accumulation exagérée de glycogène et d'hypoglycémie, car le foie ne peut pas fournir de glucose à la circulation. Les patients atteints de cette condition ont des modifications de croissance.
- La maladie de Pompe ou de type II est due à un déficit α-déficit (1 → 4) -glucano 6-glycosyltransféras dans le foie, le cœur et les muscles squelettiques. Cette maladie, comme Andersen ou Type IV, est mortelle avant les deux années de vie.
- La maladie de Mcardle ou de type V a un déficit musculaire phosphorylase et s'accompagne d'une faiblesse musculaire, d'une diminution de la tolérance à l'exercice, d'une accumulation anormale de glycogène musculaire et d'une absence de lactate pendant l'exercice pendant l'exercice.
Les références
- Bhattacharya, k. (2015). Investigation et gestion des maladies hépatiques de stockage du glycogène. Pédiatrie translationnelle, 4(3), 240-248.
- Dagli, un., Sentner, C., & Weinstein, D. (2016). Maladie de stockage du glycogène de type III. Revues de gènes, 1-16.
- Guyton, un., & Hall, J. (2006). Manuel de physiologie médicale (11th ed.). Elsevier Inc.
- Mathews, C., Van Holde, K., & Ahern, k. (2000). Biochimie (3e érigé.). San Francisco, Californie: Pearson.
- McKiernan, P. (2017). Pathobiologie du désir de stockage du glycogène hépatique. Curr Pathobiol Rep.
- Murray, R., Bender, D., Botham, K., Kennelly, P., Rodwell, V., & Weil, P. (2009). Biochimie illustrée de Harper (28e Ed.). McGraw-Hill Medical.
- Nelson, D. L., & Cox, M. M. (2009). Principes de lehninger de la biochimie. Éditions Omega (5e Ed.).
- Rawn, J. D. (1998). Biochimie. Burlington, Massachusetts: Neil Patterson Publishers.
- Tarnopolsky, m. POUR. (2018). Myopathies liées aux troubles du métabolisme du glycogène. Neurothérapie.
- « Histoire de l'argon, structure, propriétés, utilisations
- Fonction bijective Qu'est-ce que, comment se fait-il, des exemples, des exercices »