

Prions
- 641
- 95
- Noa Da silva
Les prions sont des protéines mal pliées qui transmettent ces informations erronées à d'autres protéines et provoquent des encéphalopathies spongiformes transmissibles. Source: Wikimedia Commons Que sont les prions?
Les prions Ce sont des protéines sans génome ni acides nucléiques qui agissent comme des agents infectieux. Ils se trouvent dans la membrane cellulaire normale, seulement sous forme de protéines mal pliées et / ou avec une structure anormale à trois dimensions.
Ces protéines sont responsables de multiples maladies dégénératives et d'une mortalité très élevée qui affectent les tissus neuronaux et la structure du cerveau.
Ils sont également appelés maladies prions. Parmi les plus importants qui affectent les humains figurent Kuru, la maladie de Gerstmann-Sträussler-Scheinker, le syndrome de Creutzfeldt-Jakob et l'insomnie familiale mortelle.
Caractéristiques prions
- Les prons sont des structures de protéines présentes dans les membranes cellulaires. Ces protéines ont une forme ou une conformation modifiée [PRP (SC)].
- En ce qui concerne sa multiplication, il est réalisé par conversion des formes, comme dans le cas de la maladie tremblante. Dans cette maladie, les prions recrutent le PRP (C) (protéines prioniques de la conformation sans surveillance) pour stimuler la conversion à l'isoforme PRP (SC).
- Ces protéines inhabituelles capables de se propager, n'ont pas d'acides nucléiques. La preuve de cela est qu'ils résistent aux rayons x et aux rayonnements ultraviolets. Ces agents décomposent facilement les acides nucléiques.
- Les protéines prioniques, dont les prions (PRP) sont composées, se trouvent dans tout le corps, non seulement des êtres humains mais d'autres vertébrés sains.
- Certains chercheurs ont réussi à démontrer que, chez la souris, ces protéines activent la réparation myélinique des cellules du système nerveux périphérique. Il a également été démontré que l'absence provoque la démyélinisation de ces cellules nerveuses.
Structure à prions
Les connaissances sur la structure des prions résident principalement dans les enquêtes menées dans les bactéries Escherichia coli.
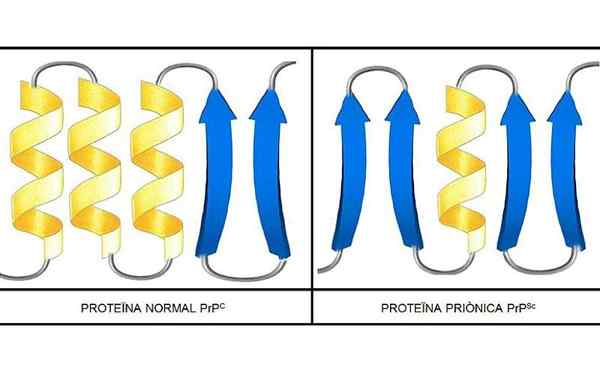
Les études ont mené que les polypeptides de la chaîne PRP (C) (normal) et PRP (SC) (infectieux) sont identiques dans la composition des acides aminés, mais diffèrent dans la conformation 3D et dans le pliage de ces.
PRP (C)
Ces prions non infectieux présents, chez l'homme, 209 acides aminés. Ils ont un lien disulfure. Sa structure est alpha-hélicoïde, ce qui signifie qu'il a des acides aminés en forme de spirale (hélices alpha) et quelques brins d'acides aminés plats (feuilles bêta).
Il peut vous servir: Sciences auxiliaires de la biologieCette protéine ne peut pas être séparée par centrifugation, ce qui implique qu'il n'est pas sédimentable. Il est facilement digéré par la large protéase du large spectre appelé protéinase k.
PRP (SC)
Il s'agit d'une protéine infectieuse qui transforme le PRP (C) en isoformes PRP (SC) infectieuses.
On sait très peu de choses sur sa structure 3D, cependant, il est connu qu'il a peu de formes hélicoïdales et de brins plus plats ou de feuilles bêta. Le changement vers l'isoforme est ce que l'on appelle l'événement fondamental des maladies de prion.
Fonctions prions
Les protéines à prion cellulaire [PRP (C)] sont situées sur la surface cellulaire d'une grande variété d'organes et de tissus. On sait très peu de choses sur les fonctions physiologiques des prions dans le corps.
Malgré cela, les expériences faites chez la souris indiquent des fonctions possibles, telles que:
Avec les récepteurs métabotropes du glutamate
Il a été démontré que le PRP (c) agit avec les récepteurs du glutamate (ionotropiques et métabotropes). Le PRP (c) participe comme récepteur des oligomères synaptotoxiques du peptide cellulaire de la surface cellulaire.
En développement embryonnaire
Chez les souris de la famille Murinae, il a été découvert que les protéines de prion PRP (c) sont exprimées quelques jours après l'implantation, en développement embryonnaire.
Cela indique qu'ils jouent un rôle pendant le développement de ces petits mammifères. L'article qui, selon les chercheurs, est lié à la régulation de la neurinogenèse (production d'axones et de dendrites de neurones).
Ils agissent également dans la croissance axonale. Ces protéines prions sont même impliquées dans le développement du circuit cérébelleux. Pour cette raison, on pense que l'absence de ces prions PRP (c) implique un retard dans le développement moteur des rongeurs.
Neuroprotecteur
Dans des études sur la surexpression du PRP (c) en raison de l'orientation des gènes, il a été constaté que l'absence de ces prions provoque des problèmes d'irrigation sanguine à certains endroits du cerveau (ischémie cérébrale aiguë).
Cela signifie que les protéines prions fonctionnent comme des neuroprotecteurs. De plus, il a été démontré que la surexpression du PRP (C) peut réduire ou améliorer les lésions causées par l'ischémie.
Système nerveux périphérique
Récemment, la fonction physiologique du PRP (C) a été découverte dans le maintien de la myéline périphérique.
Peut vous servir: dystrophine: caractéristiques, structure et fonctionsAu cours d'une étude de laboratoire, il a été découvert qu'en l'absence de protéine prion, les souris de laboratoire ont développé des carences nerveuses qui transfèrent des informations du cerveau et de la moelle épinière, dans ce qu'on appelle la neuropathie périphérique.
Mort cellulaire
Il y a des protéines prions et sont situées dans d'autres parties du corps autre que le cerveau.
Les fonctions de ces protéines consistent à initier, réguler et / ou contrôler la mort cellulaire, lorsque le corps est attaqué (par vulons, par exemple), empêchant ainsi la propagation du pathogène.
Cette fonction particulière de ces protéines fait réfléchir aux chercheurs de l'importance possible des prions non infectieux dans la lutte contre les agents pathogènes.
Memoire à long terme
Une étude menée au Stowers Institute, au Missouri, EE. Uu., Il a montré que les prions PRP peuvent avoir une fonction dans le maintien de la mémoire à long terme.
L'étude a révélé que certaines protéines prions peuvent être contrôlées pour fonctionner sur le maintien des fonctions physiologiques de la mémoire à long terme.
Renouveau des cellules mère
Une étude sur les protéines prions qui sont exprimées dans les cellules tissulaires des tiges, a révélé que toutes ces cellules souches (hématopoïétique), expriment des protéines prions dans leur membrane cellulaire. On pense donc qu'ils participent au processus complexe et très important de renouvellement cellulaire.
Maladies causées par les prions
Les maladies de prion les plus courantes sont:
Creutzfeldt-Jakob (ECJ) maladie (ECJ)
Considéré comme la maladie de prion la plus courante chez l'homme, c'est une pathologie cosmopolite, c'est-à-dire la distribution mondiale. Cela peut se produire l'héritage (famille), sporadique ou infectieux.
Maladie de Gerstmann-Sträussler-Scheinker
Il s'agit d'une maladie causée par les prions dans un processus encéphalique hérité infectieux ou autosomique dominant. La maladie se manifeste chez les personnes de 40 à 60 ans.
Prionopathie avec sensibilité variable aux protéases
C'est une maladie très rare, au point que sa gamme d'occurrence est de 2 à 3 cas pour 100 millions d'habitants. La pathologie est similaire à la maladie de Gerstmann-Sträussler-Scheinker.
Insomnie létale
C'est une maladie héréditaire ou familière, bien qu'elle puisse également se produire sporadiquement. On sait que la maladie est due à une mutation héréditaire ou autosomique dominante.
Peut vous servir: espèces endémiquesKuru
Cette maladie à prion n'a été détectée que chez les habitants de Papouasie-Nouvelle-Guinée. C'est une maladie liée au cannibalisme et à la tradition culturelle du rite duel pour les morts, où ces gens mangent le cerveau humain.
Maladies chez les animaux
Parmi les pathologies produites par le prion chez les animaux est l'encéphalopathie spongiforme bovine. Cette maladie a fait des ravages en Europe, en santé publique, celle des animaux et de l'économie des pays touchés.
D'autres maladies chez les animaux sont le scracyre, l'encéphalopathie transmissible des visons, la maladie d'usure chronique (chez le cerf) et l'encéphalopathie spongiforme féline.
Ces maladies, comme celles présentées chez l'homme, n'ont pas de traitement efficace, donc la prévention est fondamentale, surtout après les contagios chez l'homme qui s'est produit en raison de la consommation de viande de vaches infectées.
Traitements
À ce jour, aucun remède contre les maladies de prion n'est connu. Le traitement est symptomatique. Il est conseillé aux patients de planifier des soins palliatifs et une analyse génétique et des conseils pour les membres de la famille sont recommandés.
Une grande variété de médicaments a été testée chez des patients atteints de maladies à prions, telles que l'antiviral, antitumoral.
Cependant, il n'y a actuellement aucune preuve qui indique que certains de ces derniers diminuent les symptômes ou améliorent la survie des malades.
La prévention
Les prons résistent à une variété de changements physiques et chimiques. Cependant, différentes techniques sont utilisées pour éviter la pollution des patients atteints d'instruments chirurgicaux contaminés.
Parmi les techniques les plus couramment utilisées, il y a stériliser l'équipement dans une autoclave à 132 ° C pendant une heure, puis submerger les instruments dans l'hydroxyde de sodium pendant au moins une heure de plus.
D'un autre côté, l'Organisation mondiale de la santé (OMS) a développé des mesures pour éviter la propagation des maladies à prions. Cette organisation établit des règles pour la gestion des tissus interdits ou potentiellement risqués tels que: les yeux, le cerveau, l'intestin, les amygdales et la moelle épinière.
Les références
- Prion, agent infectieux. Récupéré de Britannica.com.
- Qu'est-ce qu'un prion? Récupéré de scientifican.com.
- Prion. Récupéré de.Wikipédia.org